Data Carpentry -for- Ecologists
Teaching the tools to get computers to do cool science

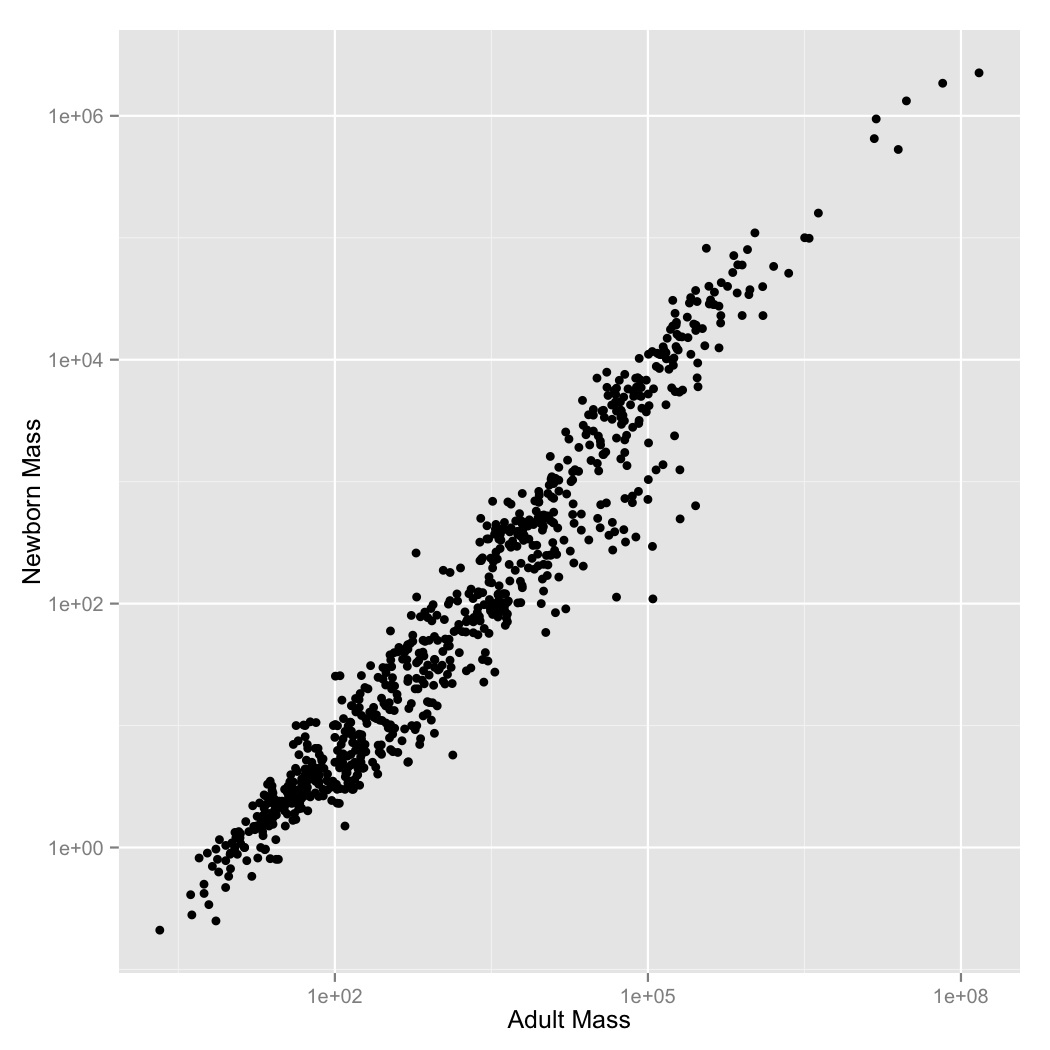
It makes sense that larger organisms have larger offspring, but what the mathematical form of this relationship should be is unclear. Let’s look at the problem empirically for mammals.
Download some
mammal life history data
from the web. You can do this either directly in the program using read.csv()
or download the file to your computer using your browser, save it in the data
subdirectory, and import it from there. It is tab delimited so you’ll want to
use sep = "\t" as an optional argument when calling read.csv(). The \t is
how we indicate a tab character to R (and most other programming languages).
When you import the data there are some extra blank lines at
the end of this file. Get rid of them by using the optional read.csv()
argument nrows = 1440 to select the valid 1440 rows.
Missing data in this file is specified by -999 and -999.00. Tell R that
these are null values using the optional read.csv() argument,
na.strings = c("-999", "-999.00"). This will stop them from being plotted.
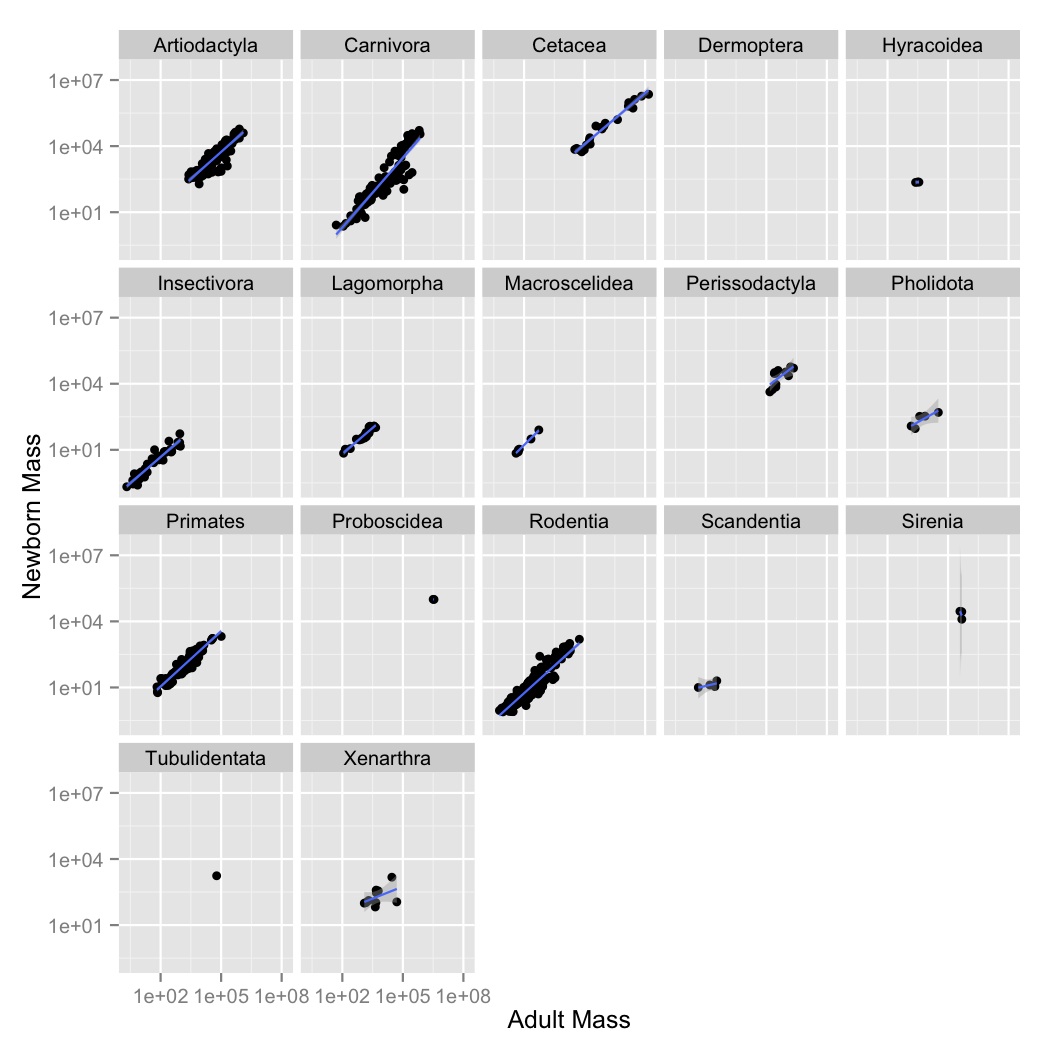
facet_wrap to
create subplot for each order.geom_smooth to fit a linear model to each order. You can do this using the
optional argument method = "lm" in geom_smooth.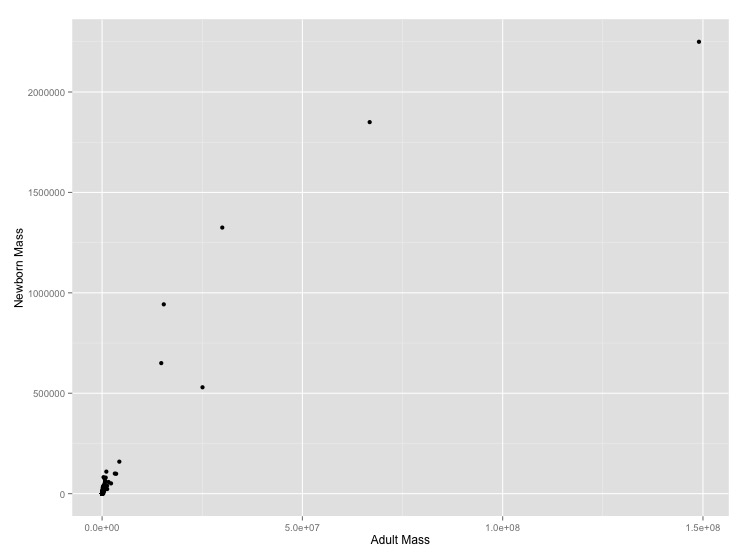{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}